Molecular Docking for potential natural drug candidates for SARS-CoV-2
SARS-CoV-2: Potential Natural Drug Candidates
After the declaration of the highly infectious disease COVID-19 (SARS- CoV-2), by the World Health Organization (WHO) in March 2020, molecular dockings approaches have gained much importance in recent years. Thus, several studies employing in silico methods, have been published and focused on identifying compounds with binding potential to proteins crucial for the functioning of the SARS-CoV-2 virus.
Key receptors of COVID-19
Previous research has tested different natural substances and drugs with potential antiviral or antiviral effect already known in the literature, for their binding to SARS-CoV-2 proteins. Of all, hesperidin was the most suitable to bind to the “spike”. By superimposing the ACE2-receptor binding domain (RBD) complex on the hesperidin-RBD complex, a clear overlap of hesperidin with the ACE2 interface is observed, which suggests that hesperidin may disrupt the interaction of ACE2 with RBD. A second theoretical site of low energy binding of hesperidin with SARS-CoV-2 is the main protease that allows the processing of the first proteins transferred from the viral genome-pp1a and pp1ab-into functional proteins in the host cell. Other findings revealed that the cell entry of SARS-CoV-2 depends on the binding of the viral spike (S) protein to the cellular receptor angiotensin-converting enzyme2 (ACE2). After the cellular entry, both the viral 3-chymotrypsin-like protease and papain-like protease (PLpro) further cleave and process the viral polyproteins to produce four essential structural proteins: the S protein, nucleocapsid (N) protein, membrane (M) protein, and envelope (E) protein, which are needed to compose a complete viral particle.
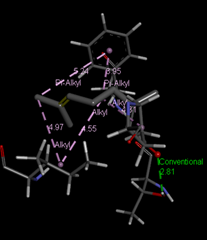
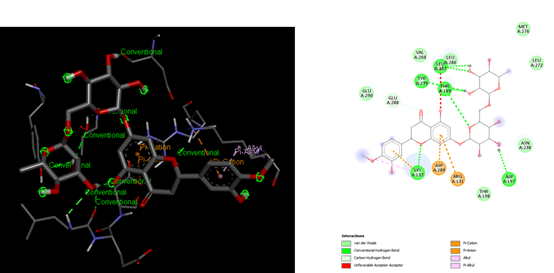
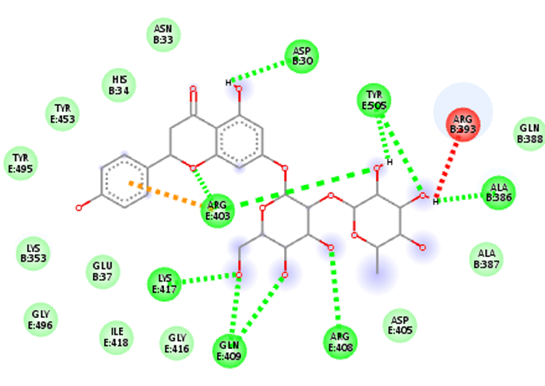
At SinodosChemistry.com we have been performing molecular dockings between natural compounds found in various plants (e.g. Citrus Fruits), such as hesperidin, citral, and naringenin, and different receptors associated with SARS-CoV-2. In particular, we explored the binding potential of these natural ligands to key targets such as Mpro, Spro, 3CLpro, and the receptor-binding domain (RBD)/ACE2 complex of the spike (S) protein, which are linked to the dynamics of COVID-19.
Natural substances with potential antiviral or antiviral effect
The evaluation of repositioned drugs for treating COVID-19 is ongoing, and concerns about severe side effects highlight the need for safe and effective treatment options. Therefore, natural products, with a proven track record in contributing to treatments against various viruses, emerge as a valuable resource in the development of COVID-19 treatments. Despite the fact that developing bioactive natural products against a specific disease, such as COVID-19, is faster than developing a vaccine, it is still a difficult process due to the diversity of natural metabolites, their chemical complexity, and the extraction process. To overcome these challenges, in silico analysis by molecular docking is a time-saving and less expensive method for conducting virtual screening for bioactive compounds
Methodology and Key findings
Our research for MVS Pharma, has shown that among the selected natural compounds, Naringenin and Hesperidin have the best binding affinities. The ligand Naringenin exhibited the highest binding affinity of -8.3 kcal/mol with the 3CLpro receptor, suggesting its potential as a drug candidate. While the ligand Hesperidin, evaluated against both 3CLpro and RBD-ACE2, demonstrated binding affinities of -8.0 kcal/mol. These findings suggest the potential of these natural products as candidates for the development of COVID-19 treatments. However, the exploration of their chemical interactions with key viral receptors is still undergoing. Indeed, our capabilities enable us to lead cutting-edge research on new bioactive compounds and validate their interactions with the key receptors associated with SARS-CoV-2. As part of our approach, we use computational tools such as UCSF Chimera and AutoDock Vina for molecular docking preparation and simulation, as well as tools like BIOVIA Discovery Studio Visualizer to visualize and analyze molecular docking results.
Contact us to discuss your molecular docking, or drug design needs!