Polymer conformations
A well discussed subject in polymer science and technology is the field of chain conformations. For decades now, scientists have been suggesting conformation and configuration models that explain partially or completely the behavior of single and grouped polymeric chains. Both topics can be discussed on a statistical, thermodynamic, or mechanical basis since both conformations and configuration are affected by such factors.
Perhaps the simplest example when discussing conformations of a polymer chain is the use of a segment of a polyethylene chain; in that case one can study the energy of the segment [or just an ethylene’s molecule] as a function of the relative positioning of all substituent atoms [hydrogen] in the two carbon atoms studied. When two hydrogens at different carbon atoms are covering each other and thus are at their closest distance, the total energy of the molecule is significantly increased, and that conformation is hard to appear in reality. This high energy, unfavorable structure is termed as ‘ecliptic’. When these two hydrogens are found at total opposite positions to each other, the energy is minimized due to electrostatic repulsions minimization and the structure is termed ‘trans’. Trans conformations are the most stable conformations and the most favored for observation during conformation investigations. Various conformation also appear that fall within the two limits of trans and ecliptic transformations, depending on the hydrogen – hydrogen angle in the two carbon atoms. This is very well depicted in figures 1 and 2 for butane. In this case, the investigation focuses on the CH3 – CH3 interactions although hydrogen – hydrogen and CH3- hydrogen interactions also play a significant part. When one tries to predict the total energy of each butane conformation, all interactions should be considered and not only the methyl – methyl ones.
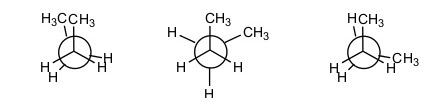
Figure 1: Butane conformations
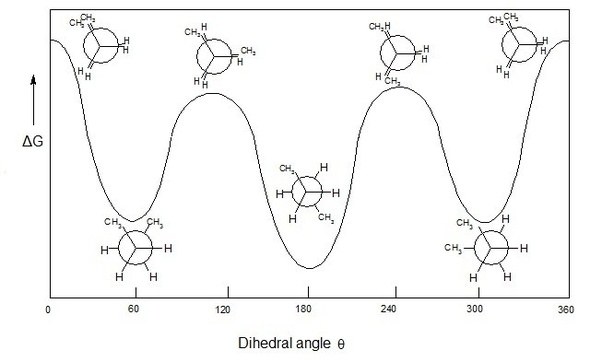
Figure 2: Butane conformational energy diagram [source: QUORA]
One should perceive the polymeric chain, especially when in a solution, as a free moving chain of literally countless conformation chances. Imagine that in reality, the angles between substituents in a carbon – carbon bond are continuous and thus the combinations of conformations in a polymeric chain become infinite. If for example in a C-C bond with three hydrogens in each atom [Ethane] there only three distinct positions for the hydrogen atoms, all possible conformations will be 32. For a short chain with just ten atoms, the possible conformations would be 102. However, with N distinct positions for the hydrogen atoms the combinations would explode to 10N, and this is the real case.
One could visually identify and understand this description by watching the following video of an average size Poly ethylene chain. In this video, Molecular Dynamics have been conducted on a single PE chain at 300K and at constant temperature. The total energy of the system is also calculated and graphed simultaneously so that one can correlate the changes with the structural changes in the chain. Constant hydrogen motion along with constant bond vibrations are evident and allow for deeper understanding of a real polymeric chain behavior. We rotate the chain periodically to show some carbon atoms in ‘a row’ so that the spectator can identify that there are no distinct spots for hydrogen atoms but a continuous range of motion around the carbon atoms. This video also allows for an initial view on configuration of chains, a topic that will be addressed in detail separately. Similar observations can also be made by watching the PVC based investigation, under the same conditions.
These two videos reveal only a part of the power and the potential of modern computational tools for the investigation of polymer chain conformations and configurations. More investigations and model building training videos will be published soon at SinodosChemistry.com. Computational tools allow not only for accurate calculation of properties at atomistic level such as bond lengths, bond angles, conformation energies, etc but also provide visual understanding of the physical and chemical phenomena taking place during a physical or chemical process. Investigations of polymeric molecules with larger substituents will reveal the importance of stereo prohibitions and energetically unfavored conformations. The situation will become more perplexed when one investigates larger systems like multiple chains of different sizes; entanglement and parallel orientation of segments of polymer chains will be observed, and reasons behind these observations will become clear.
One of the greatest aids that computational tools offer in conformational investigations is the ability to calculate total energy of each conformation for the chain or for a part of the chain. Recording and evaluating energetic changes as a function of atoms’ coordinates provide both accurate energy diagrams like the one predicted in figure 2, but also offer predictive analytical tools for the prediction of complex, heterogeneous chains. In bottom line, conformational investigations are a key to understanding polymer energy and behavior in solution. Understanding the unlimited combinations of atom positions and their effect on molecular energy can be used in combination with thermodynamic and statistical principles for a complete description of their chemical and physical structure.
Each conformation that can be derived as a ‘snapshot’ during a molecular dynamics investigation like the one in the PVC video, is characterized by a unique energy. Gathering a sample of these energies by collecting snapshots at given or random time intervals leads to an energy distribution [conformation distribution] diagram that reveals the most possible conformations for appearance when in solution. Averaging these chances of appearance [as a weight factor] with the respective energies, leads to an average [observed] energy for a polymeric chain that is commonly used for steady state or equilibrated processes. Dynamic values can be used to explain kinetics but the average values will be used when thermodynamic predictions are required. More works will follow on this crucial topic in the weeks to come.