Carbon Monoxide removal by Ionic Liquids: A theoretical investigation

Carbon Monoxide is one of the most toxic compounds known to man. It can be lethal at low concentrations and it is very difficult (if not impossible) to detect since it is odorless, colorless and of no taste. Its detection is based on sensors and detectors that interact with it and this interaction provokes a change of a measurable property such as an electronic, a physical, or a chemical one. Carbon Monoxide has attracted significant amount of research in the fields of sensors, detectors and removal approaches. Regarding sensors and detectors, desired properties include fast, reliable detection at low concentrations and a unique selective mechanism (one that doesn’t mistake another molecule for Carbon Monoxide). When removal approaches are discussed, the desired attributes depend on the mechanism but they mostly include adsorption/ absorption capacity and kinetics, adsorbent/ absorbent regeneration costs, life cycle, membrane costs and selectivity and others.
Our client has asked us to predict optimum sensor and detector media for Carbon Monoxide even at very low concentrations. Our work focused on first comparing three different computational chemistry approaches on confirmation of known experimental results and then using the most efficient of them to predict novel adsorbents/ adsorbents for Carbon Monoxide capture. Computational chemistry approaches tested included Ab Initio, Density Functional Theory, Semi Empirical field and MM+ field. Ab Initio and Density Functional Theory (DFT) were expected to have advantages over the other two since the investigated systems are quite novel (Ionic Liquids) and no optimized data sets exist for such compounds and their mixtures. Computational chemistry approaches were confirmed on the basis of existing experimental results on 1-hexyl 3- methyl imidazolium chlorocuprate use as an absorbent for Carbon Monoxide. Other Ionic Liquids had also been under research but with no definite results. Our primary task was to construct the 1-hexyl 3- methyl imidazolium chlorocuprate model and set up the different computational chemistry simulations. The philosophy is one of known validity; initially the molecular (and atomic/ ionic where required) energies were calculated based on the optimized molecular models. Then, the absorption systems were prepared and the energies of the systems in equilibrium were calculated. The absorption energy was then derived by equation 1:
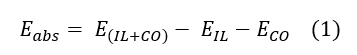
Where Eabs is the energy of absorption, E(IL+CO) is the energy of the Carbon Monoxide – Ionic Liquid complex, EIL is the energy of 1-hexyl 3- methyl imidazolium chlorocuprate and ECO is the energy of Carbon Monoxide molecule.
All simulations were run in HyperChem, HyperCube Inc molecular modelling software. Ab Initio 6-21G basis set and DFT pvdZ basis sets were employed, whereas for the Semi Empirical and MM+ different fields were used and predictions were averaged. It was concluded that both Ab Initio and Density Functional Theory (DFT) predicted the experimental energy of absorption within 10% error that can be attributed to a number of factors that include impurities in the experimental setup, stirring methodology, modifications and control of the experimental process and other. Semi empirical and MM+ fields exhibited wide fluctuations in the order of 50-70%. These inabilities of Semi Empirical and MM+ approaches to model Carbon Monoxide absorption on Ionic Liquids were expected due to the novel nature of the Ionic Liquid compound. Semi empirical and MM+ approaches require known data on similar (or the same ideally) chemical environments) that did not exist in this task. On the other hand, Ab Initio and Density Functional Theory (DFT) do not require any assumptions or knowledge of the chemical environment under investigation and thus are ideal for the study of such novel systems.
Having confirmed our ability to accurately predict the absorption of Carbon Monoxide on 1-hexyl 3- methyl imidazolium chlorocuprate we continued by examining the interactions between Carbon Monoxide and 30 Ionic Liquids that were chosen on the basis of their expected dissolution kinetics and suspected optimum cations and anions from our previous works. Our continuously growing database of Ionic Liquid properties was of great help in this task; our optimization algorithms have led us to the group of 30 Ionic Liquids for this investigation. In this second phase, we successfully identified 3 Ionic Liquids with very high affinity towards Carbon Monoxide (large negative absorption energy) and another 4 that can lead to controlled non reactive absorption of Carbon Monoxide on Ionic Liquids. Non-reactive absorption can be of great importance since it allows for low regeneration costs of the Ionic Liquid; this is due to a sterically hindering effect of cations of Ionic Liquids that prohibit the reaction between the acidic (in Carbon Monoxide) and basic (in Ionic Liquid anion) centers.
In summary, we employed Computational Chemistry approaches to predict optimum media for interaction with Carbon Monoxide. Our methodology was first validated against the very sparse experimental data available and then was used to scatter a group of ionic liquids for that cause. Once again we were successful in prediction optimized solutions for our clients that depending on the interaction mechanism and the potential change of system properties, can be used for sensors, detectors or removal materials for Carbon Monoxide from gaseous mixtures.